2 minutes
Insights into Bacterial Genome Sequence Analysis
Today goals
- Things you should know before assembly
- Fastq format
- Fasta format
- GC- content
- Status of genome
- Step to bacterial sequencing analysis
Things you should know before assembly
Fastq format
start with @
@cc3e68c4-b53d-43da-be7e-b961113007e2 -> sequence name
ATCCGGAATCGGTTACTGTTGGGAACCTTTGC -> sequence
+ -> quality line break
#%(*))++.2/148447;7+001./18-7-,,30&*2 -> quality score
Fasta format
start with >
>tig00000001 -> sequence name
TGATAAAAGTATTCATATAATCTCCTATCATTTCAAAATTTAAT -> sequence
>tig00000002
ATATTAGTGTGTCTATTTTATGGGGCTAGGAAAGGAGGTACATT
GC-content
the percentage of nitrogenous bases on a DNA or RNA molecule that are either guanine or cytosine $$ \frac{G+C}{A+T+G+C} \times 100 \% $$
Satuts of genome

1. contig
A contig is a set of overlapping DNA segments that together represent a consensus region of DNA.
2. scaffold
To bridge the gaps between the two contigs called scaffold.
3. complete
Didn’t have fragment, chromesome is in one contig

Step to bacterial sequencing analysis

Step 1. Quality Control - Assessing the quality of TGS
1.1 Checking raw read statistics
Tools : abyss-fac
$ abyss-fac -t 1 read.fastq contig.fasta

n : Number of raw read
N50 : A value that present 50% of sorted read set

Max: longest read length
Sum: Total read length
1.2 QC report
Tools : FastQC
$ fastqc -f read.fastq -o outdir
Step 2. Trimming Filtering Data - Preprocessing of raw data (optional)
2.1 Filter raw read
Tools : filtlong
Usually used in coverage too high, trim too short reads, and want to keep raw read in between a range
2.2 Demultiplexing, Trimming adapter (barcode)
Tools : porechop, deepbinner
$ porechop -i reads.fastq -o output.fastq # adapter trimming
$ porechop -i reads.fastq -b output.fastq # demultiplexing
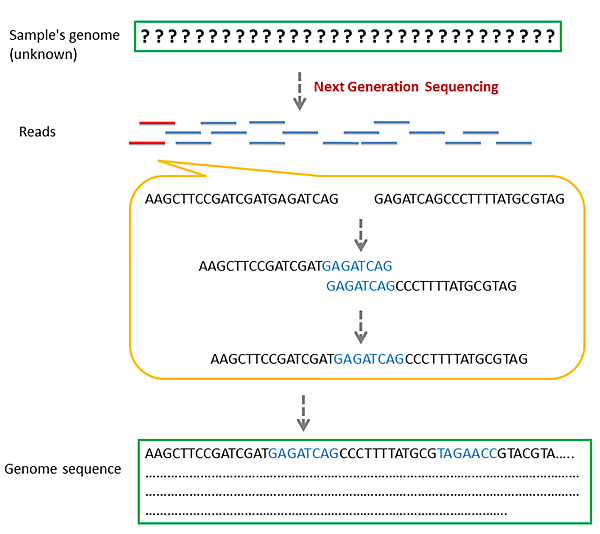
Step 3. Sequence Assembly - Long read genome assembly
3.1 De novo assembly
Tools : Canu, Unicycler, Flye, Ra
$ canu -p genomename -d outdir genomeSize=4.8m -nanopore-raw read.fastq
$ unicycler -l read.fastq -o outdir
...
Step 4. Assembly Validation
4.1 Assembly evaluation
Tools: abyss-fac, assembly-stats
- Total assembly size
- Total number of sequence
- Longest contig
- Average contig size
- N50
4.2 Assembly status graph
Tools : Bandage
Is it circular or linear ?